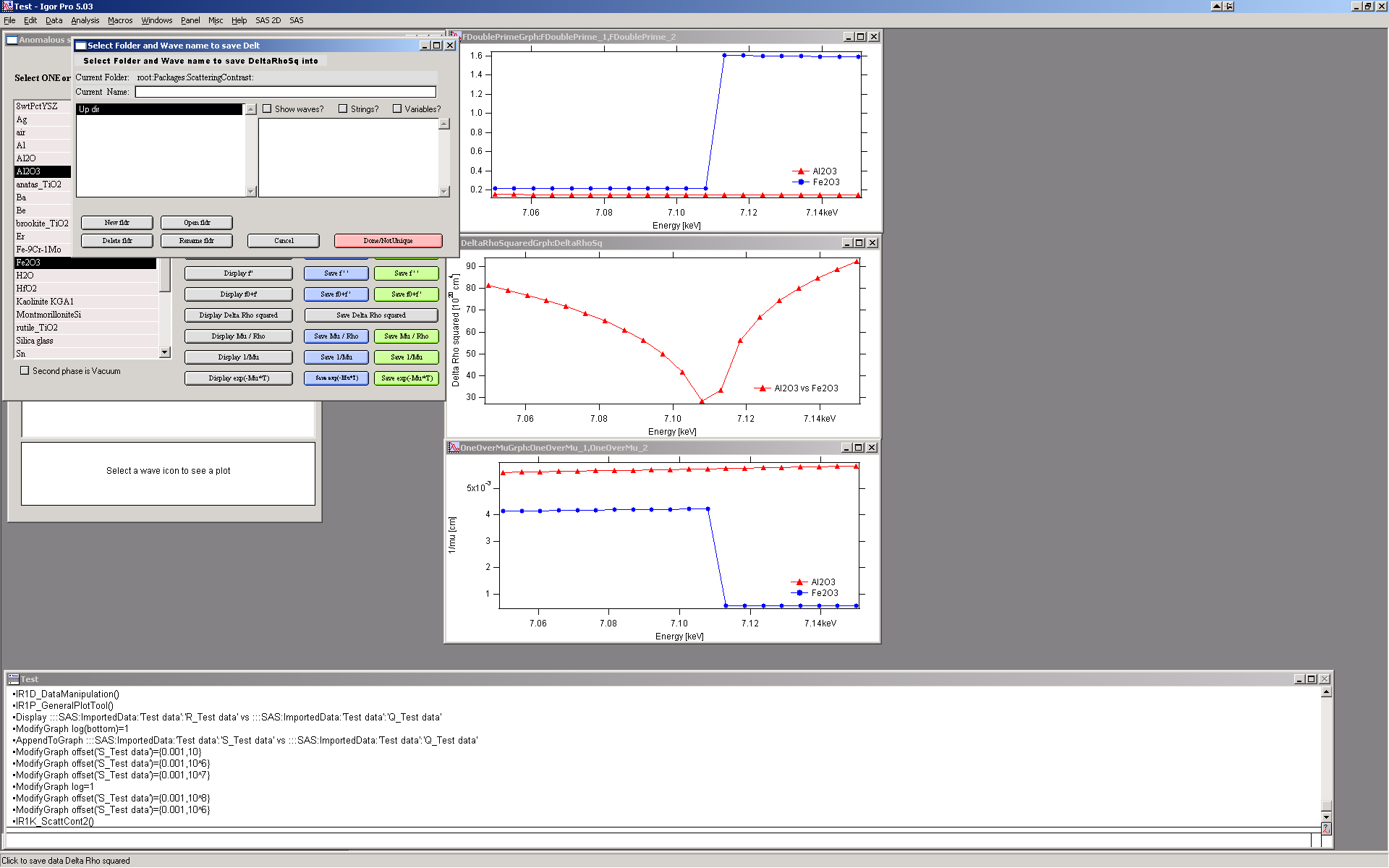
Scattering contrast calculator¶
Calculating scattering contrast for compounds is tedious but well-suited for automation. This tool calculates X-ray and neutron scattering contrast for compounds with up to 24 atoms, using known density and atomic fractions, without energy dependence. Energy-dependent anomalous scattering effects (for X-rays) can be included using the Anomalous calculator (see the button in the lower-right corner).
Basic calculator¶
Select “Scattering contrast calculator” from the SAS menu:
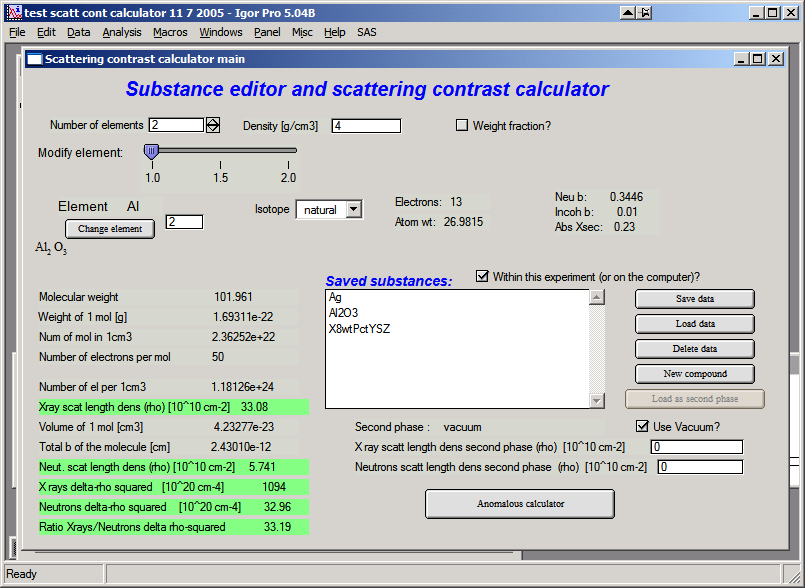
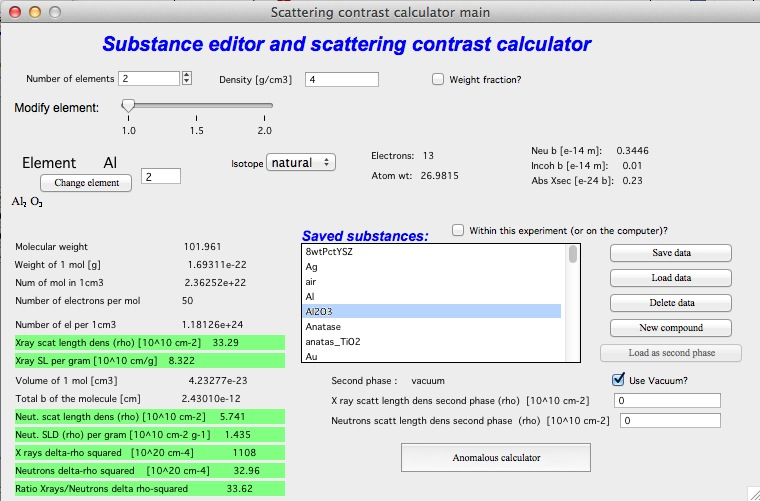
At the top, select the number of atoms in the compound, enter its density, and check the checkbox if neutron data should be shown. For example, selecting 2 atoms for Al2O3 (Corundum) with a density of 4.0 and enabling neutron results gives:
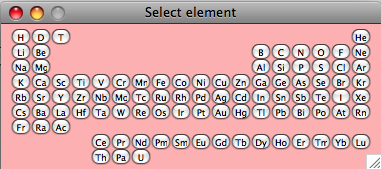
Use the slider to select each element and check its properties — amount in the formula unit, isotope, etc. Element selection is done through the Periodic Table (click “Change element”, then close the table to continue):
Most fields are filled automatically from the internal databases. The lower section of the tool shows results and intermediate calculations.
Matrix
To calculate (Δρ)² = (ρmatrix − ρscatterer)², the scattering length density of the matrix must be specified. This can be done in several ways:
Enter values directly in the provided fields.
Calculate the matrix SLD with the tool and click “Set as matrix”.
Save compound data with “Save data” and reload as matrix with “Load matrix data”.
In each case the Δρ² values are recalculated automatically. When “Use vacuum as matrix” is checked, vacuum is used and no matrix selection is available.
Saving data
Compound parameters can be saved for future use, either inside the current Igor experiment or on the computer (accessible to all Igor experiments on that machine, but not portable to other computers). Use the checkbox “Within this experiment (or on the computer)?” to select the storage location.
“Save data” — saves the current compound. Modify the name as needed and keep it within 27 characters (Igor name limit). Retain the quotes around the name.
“Load data” — loads a saved compound.
“Load matrix data” — loads a compound as the matrix only.
“New compound” — clears all settings to start a new compound definition.
Note
Loading saved data from ASCII files introduces small rounding errors that affect (Δρ)² calculations via “Load matrix data”.
From the current release, compound data are saved in the same location as the Irena macros, allowing users with limited file system privileges to use the feature.
Anomalous calculator¶
The package includes Cromer-Liberman code for calculating energy-dependent (anomalous) scattering effects. Click “Anomalous calculator” in the Substance editor panel to open the anomalous calculator:
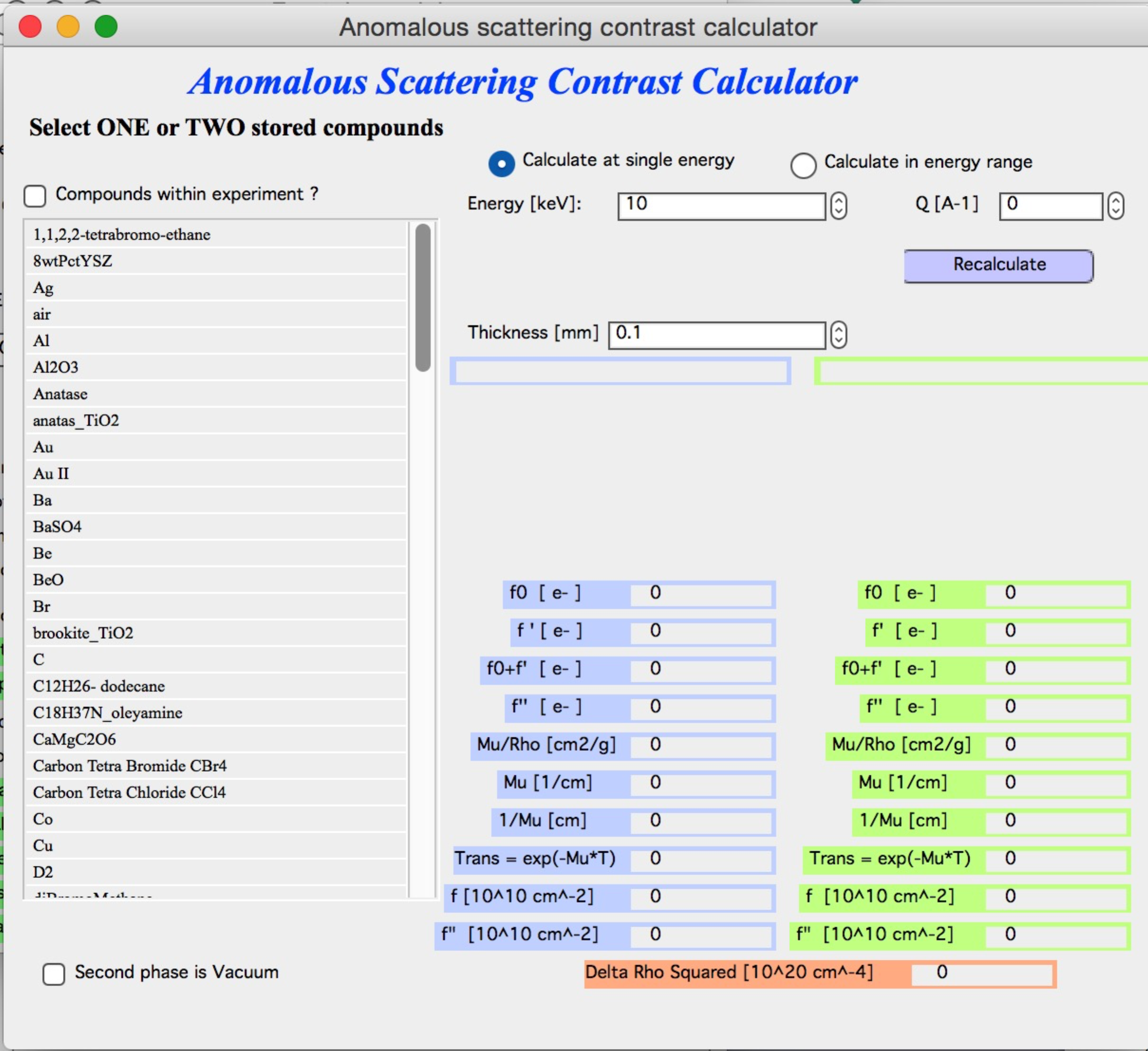
Select one or two compounds that have been created and saved in the basic scattering contrast calculator. If only one compound is selected, use vacuum as the second phase (checkbox below the compound selector). Choose whether to calculate at a single energy or over an energy range. Note that calculating over many points can take a significant amount of time.
To select two compounds, Shift-click. Enter the appropriate thickness and click “Recalculate”. Enter the Q value if results at a specific Q are needed (for small-angle scattering, assume Q = 0).
Single energy
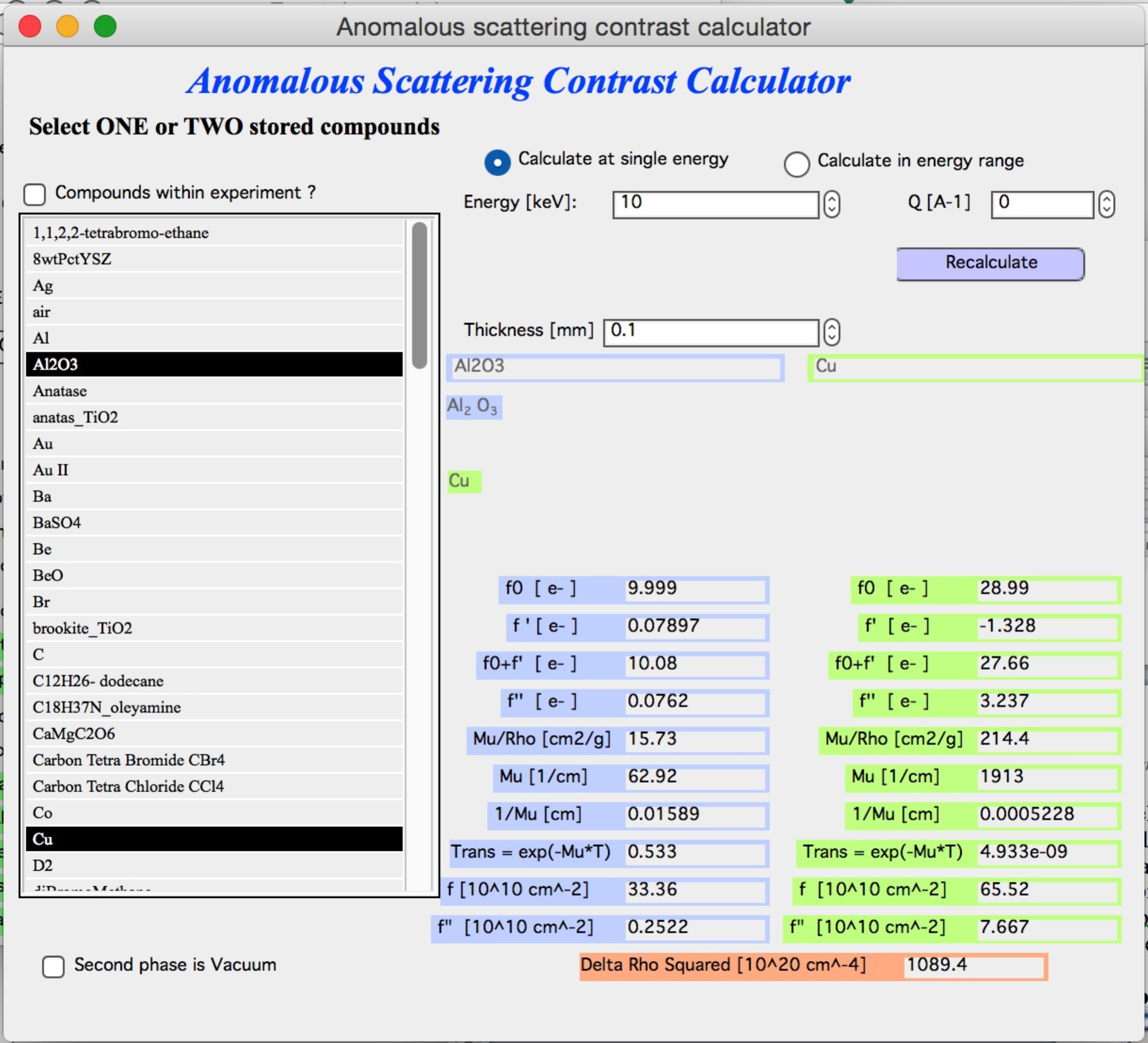
The table on the right is populated with all relevant values: f’, f”, μ, and related quantities for each compound. f’ and f” are given in two unit conventions — electrons per molecule and 1010 cm-2. The lowest value shown is (Δρ)² between the two compounds at this energy.
The line transm = exp(−μT) gives the calculated transmission of each
material at the entered thickness (in mm) and selected energy. Changing the
thickness automatically recalculates transmission — useful for estimating
required sample thickness before an experiment.
Range of energies
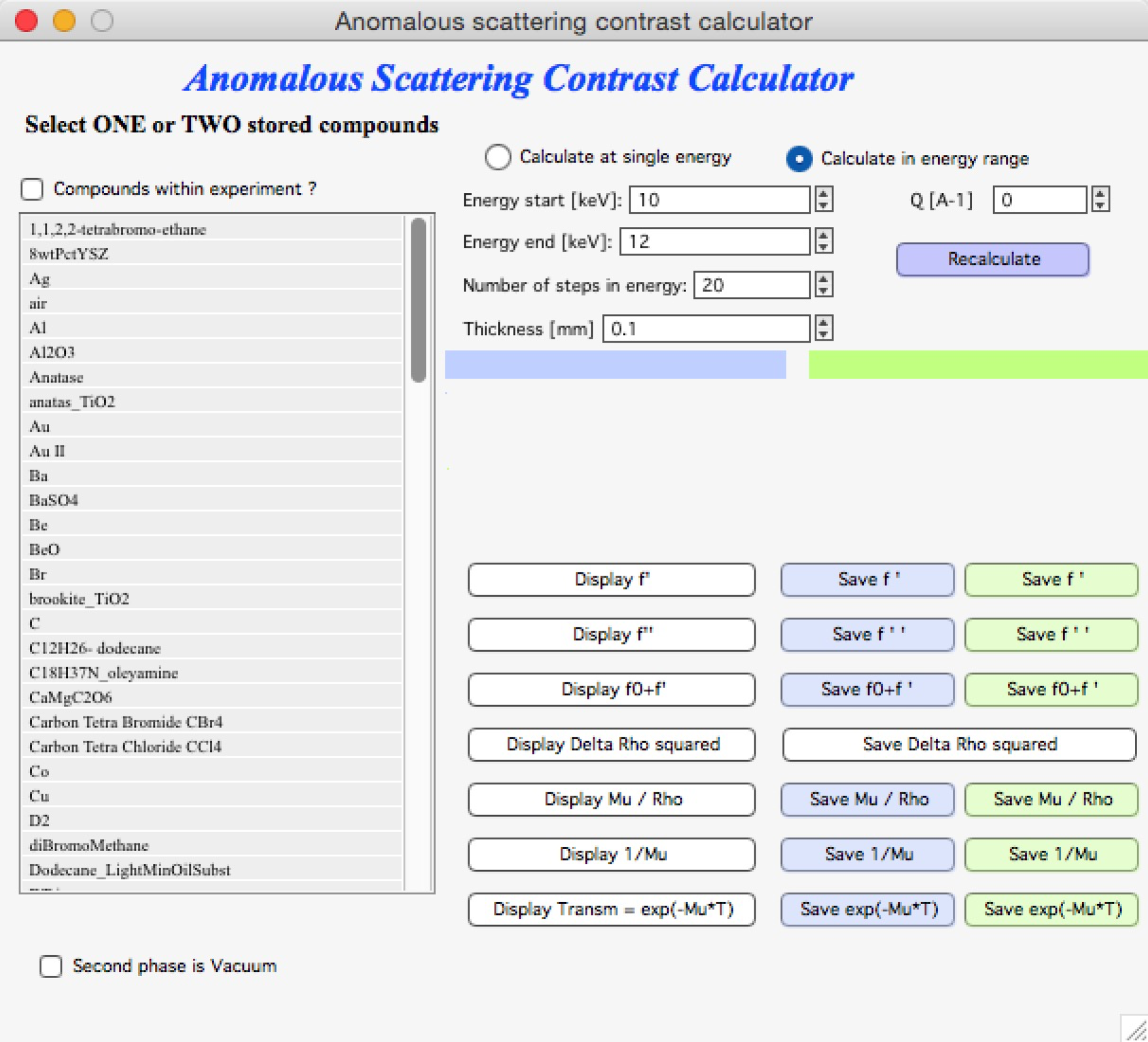
Enter the energy range, number of calculation steps (equally spaced between minimum and maximum), and other parameters, then click “Recalculate”. The “Display” buttons create graphs of the calculated parameters:
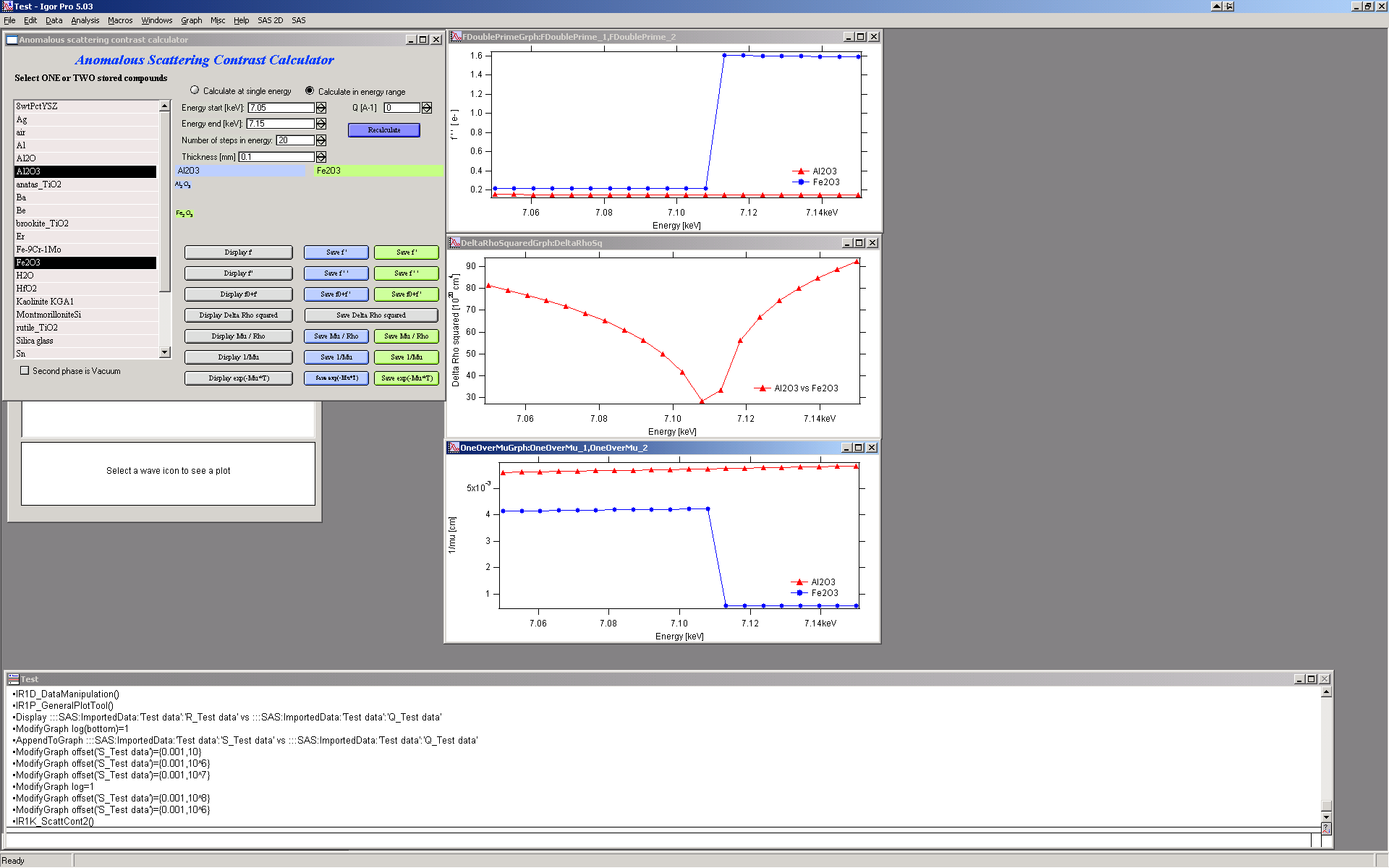
The “Save …” buttons save the result waves to an Igor folder of your choice. The data are saved with x-scaling — see the Igor Pro manual for details.